Болезнь Гоше — это наследственное заболевание из группы болезней накопления (лизосомных болезней), связанное с врожденным нарушением обмена липидов 1.
Суть заболевания
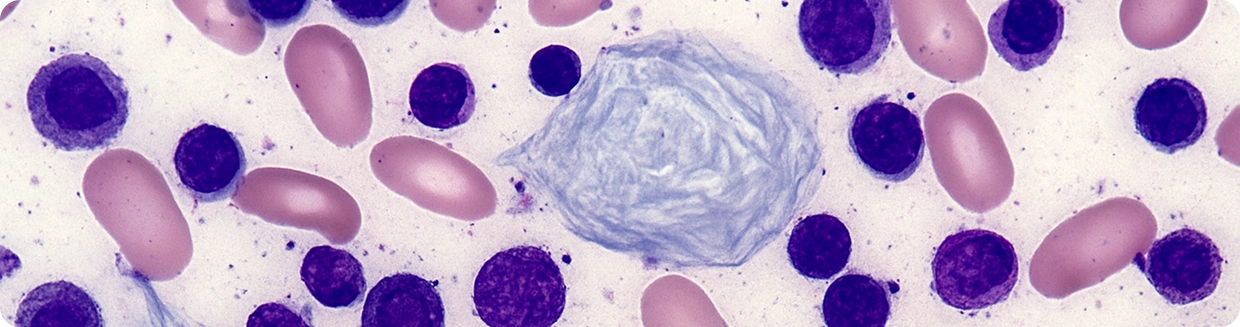
При болезни Гоше фермент β-глюкоцереброзидаза, расщепляющий внутриклеточные липиды работает недостаточно или отсутствует. В результате нерасщеплённые липиды накапливаются внутри макрофагов (так называемых «клеток Гоше»). Эти клетки постепенно откладываются в селезёнке, печени, костном мозге, а иногда и в костях и других органах, вызывая анемию, тромбоцитопению, костные и другие нарушения в организме .
Распространенность болезни Гоше в общей популяции составляет 1 случай на 40 000 новорожденных .
Классификация болезни Гоше4
I тип
- Наиболее распространенная форма в западных регионах (95% случаев)
- Нервная система не страдает, интеллектуальное развитие остается в норме
- Это единственная форма, которая хорошо поддается лечению на сегодняшний день
II тип
- Встречается очень редко; болезнь быстро прогрессирует и приводит к летальному исходу в раннем детстве
- Сопровождается тяжелыми неврологическими нарушениями (поражение ствола головного мозга)
- Существующие методы лечения не могут остановить повреждение нервной системы
III тип
- Является наиболее распространенной формой заболевания в мире, но в западных регионах встречается редко
- Сочетает в себе признаки I типа и умеренно выраженные неврологические симптомы, которые прогрессируют медленнее, чем при II типе
- Продолжительность жизни варьируется, в некоторых случаях при правильном лечении может доходить до 50 лет
Ключевые симптомы2
(для всех форм заболевания)
Диагностика5
Болезнь Гоше I типа, наиболее распространенная в западных регионах, часто остается недиагностированной в течение многих лет, так как ее симптомы могут имитировать другие заболевания. Это приводит к задержке постановки диагноза и началу лечения. При наличии сочетания гепатоспленомегалии, цитопении (анемия, лейкопения, тромбоцитопения) и/или костных изменений у пациента необходимо:
Первичное обследование
- Биохимический анализ (ферменты печени, маркеры резорбции костной ткани)
- УЗИ органов брюшной полости (особенно печени и селезенки)
Специализированная диагностика
- Генетическое тестирование (мутации в гене GBA)
- МРТ трубчатых костей (оценка поражения костной ткани, наличие гошером)
Подтверждение диагноза и направление к специалисту
- Рассмотрение возможности назначения ферментозаместительной терапии
Для специалистов здравоохранения. Информация не является рекомендацией компании Такеда, рекламой компании или ее продукции, не должна быть основанием для принятия каких-либо решений или осуществления каких-либо действий. Решение о выборе метода лечения конкретного пациента должно приниматься лечащим врачом.
Список литературы:
- Brady RO. Gaucher's disease: past, present and future. Baillieres Clin Haematol. 1997 Dec;10(4):621-34.
- Mistry PK, Lopez G, Schiffmann R, Barton NW, Weinreb NJ, Sidransky E. Gaucher disease: Progress and ongoing challenges. Mol Genet Metab. 2017 Jan-Feb;120(1-2):8-21.
- Oliveri, B., González, D., Quiroga, F., Silva, C., Rozenfeld, P. AComprehensive Study of Bone Manifestations in Adult Gaucher DiseaseType 1 Patients in Argentina. Calcif Tissue Int 104, 650-657 (2019).
- National Gaucher Foundation, https://gco.iarc.who.int/media/globocan/factsheets/cancers/33-hodgkin-lymphoma-fact-sheet.pdf (last access: September 2025).
- Mistry PK, Sadan S, Yang R, Yee J, Yang M. Consequences of diagnostic delays in type 1 Gaucher disease: the need for greater awareness among hematologists-oncologists and an opportunity for early diagnosis and intervention. Am J Hematol. 2007 Aug;82(8):697-701
Информация предназначена для специалистов здравоохранения.
VV-MEDMAT-126748/ОКТЯБРЬ 2025
Approved in Marv: OCTOBER 2025/Valid till OCTOBER 2027